

Abstract
Introduction: Lupus nephritis (LN) is associated with a hypercoagulable state and an increased risk for thrombosis. Glomerular microthrombosis is detected in approximately 33% of LN patients. The incidence of thrombotic disease in patients with LN is 1.35 to 6.2 times higher than in other systemic lupus erythematosus (SLE) patients. While the immune complex is thought to elicit most forms of injury in LN, thromboembolic complications may be another important cause of renal injury and kidney dysfunction. However, the mechanisms specific to LN that promote a hypercoagulable state have not yet been identified. Antiphospholipid antibodies (aPLs) have been found to severely affect the pathophysiology of LN. But several authors have reported that the presence of aPLs is not sufficient for thrombus formation in vivo. Despite the clinical efficacy of the anti-inflammatory and immunosuppressive properties of glucocorticoid (GC) in treating LN, increased risk of thrombosis has been observed in GC users. Therefore, further studies should aim at finding other appropriate targets and effective treatments to correct coagulation abnormalities in LN. Recent studies have shown that phosphatidylserine (PS), a membrane constituent, plays an important role in thrombosis. However, the procoagulant role of PS in LN is not fully understood. Our objective was to elucidate the effects of PS exposure on microparticles (MPs) and their originating cells in LN.
Methods: LN patients (n = 40) were divided into two groups, defined as active LN (aLN) and inactive LN (iLN), and compared with non-LN SLE patients (n = 20) and controls (n = 20). Patients with aLN were those with impaired kidney function with proteinuria (>0.5 g/day), an active urine sediment, or kidney biopsy with active glomerulonephritis. Inactive LN patients had decreased proteinuria levels (<0.5 g/day) and inactive urinary sediment with stable kidney function. PS exposure on MPs and MP-origin cells was quantified by lactadherin, and resulting procoagulant activity (PCA) was assessed with coagulation function assays. Fibrin production was determined by turbidity. Phosphatidylserine exposure and fibrin strands were observed using confocal microscopy. To analyze the role of glucocorticoids in MPs generation, pooled MPs suspensions were prepared from paired samples of each patient group before and after glucocorticoid treatment.
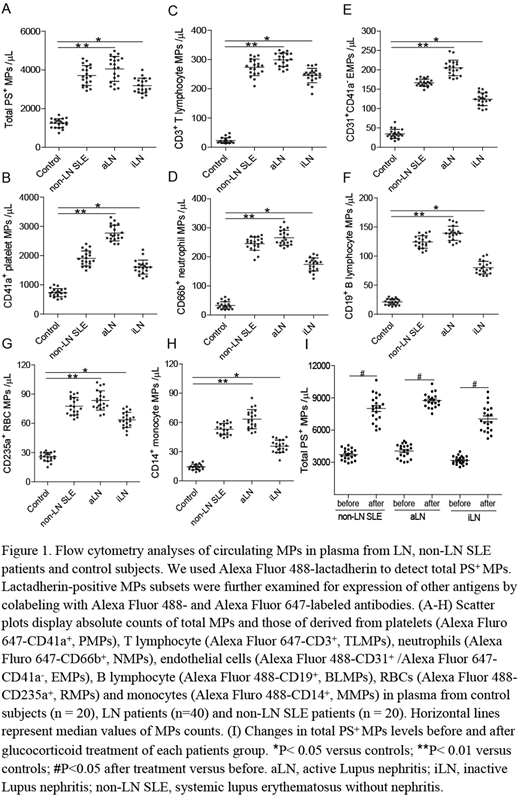
Results:We found that LN patients had high levels of PS+ MPs, blood cells (BCs) and cultured endothelial cells (ECs) by flow cytometry. Circulating levels of all MP subsets were significantly higher in aLN, with the largest increases seen in MPs derived from platelets (CD41a+, PMPs), T lymphocytes (CD3+, TLMPs) and neutrophils (CD66b+, NMPs) compared to controls (all P<0.01, Figure 1A-H). Furthermore, the aLN patients had greatly elevated PS+ MPs compared with iLN. In a paired sampling study, total PS+ MPs increased significantly after glucocorticoid treatment (all P<0.05, Figure 1I). The percentage of PS+ BCs was also markedly increased in aLN, primarily in lymphocytes (32.1 ± 2.6%) followed by neutrophils (26.4 ± 1.2%), monocytes (12.5 ± 0.8%), platelets (11.3 ± 0.4%) and erythrocytes (6.2 ± 0.3%). In addition, circulating PS+ MPs and MP-origin cells in LN, especially in active stage, dramatically shortened coagulation time and increased FXa/thrombin generation and fibrin formation. Blocking PS with lactadherin prolonged coagulation time, inhibited the PCA over 80% and reduced fibrin formation. More importantly, confocal microscopy images showed fibrin strands formed on MPs and ECs in the same regions that bound lactadherin, and showed FVa/Xa costaining. Lastly, the percentage of PS+ cells significantly correlated with disease severities (proteinuria, serum creatinine, SLEDAI and urinary albumin) of LN patients.
Conclusions:The current findings suggested that these activated or injured cells may contribute to the hypercoagulability of LN through PS exposure and MPs release. Our results enable us to better understand the developing trend of the thrombosis in LN. Blockade of PS prior to thrombus formation might be a novel therapeutic approach in these patients.
No relevant conflicts of interest to declare.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal